

For decades, personalised cancer therapy has seemed like a compelling idea that never quite translated into routine practice. Every tumour is different, so why should treatment be the same? Yet, most cancers are still managed with standardised regimens. A new clinical trial may finally begin to change that. By matching therapies to each patient’s tumour DNA – and adjusting drug combinations and doses accordingly – the study shows that truly personalised cancer therapy is feasible and effective. It is the clearest clinical demonstration yet of a personalised model of cancer care that many clinicians have long believed in, but until now had little evidence to support.
The One-Size-Fits-All Problem in Oncology
While precision medicine has improved how we choose drugs, it has been far less successful at personalising how those drugs are actually used, especially when it comes to dosing and combinations. These two limiting factors help explain why most precision therapies still rely on fixed regimens and why a different clinical trial logic is needed.
The first bottleneck lies in something so routine it is rarely questioned: how cancer drugs are dosed. This dosage decision is made at the very start of clinical development. In phase I oncology trials, doses are escalated between patients until they identify a maximum tolerated dose (MTD), the highest dose that produces an acceptable rate of serious side effects. That MTD then shapes the recommended phase II dose (RP2D), and sometimes the optimal biologic dose (OBD) for all patients in later trials and, eventually, routine practice.
The problem is that the MTD can be derived from very small numbers of patients (as few as six) and is driven by early toxicities, typically within the first treatment cycle (around four weeks). The derived MTD is then applied to everyone, even though patients may not process drugs the same way. Studies already show that drug metabolism differs by age, sex, body weight, race/ethnicity, organ function and metabolic or genetic variations (Figure 1). A fixed dose, therefore, leaves some patients undertreated (too little drug, blunted efficacy), while pushing others into overtreatment (too much drug, therapy toxicity).
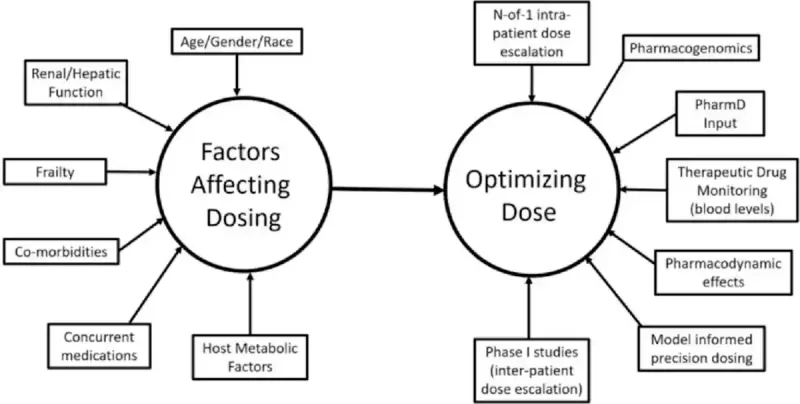
Figure 1. Cancer drug metabolism and dosing are influenced by many patient-specific factors, including age, sex, race, organ function, frailty, co-morbidities, metabolism and other medications. Personalised dosing strategies, such as starting at lower doses and adjusting within each patient (“N-of-1” intra-patient), aim to deliver the safest and most effective dose for each individual. Source: Nikanjam et al. (2023), European Journal of Cancer.
Dosing is just one flaw in the one-size-fits-all model. A second bottleneck lies in the fact that cancers themselves are not uniform. Advances in gene sequencing have uncovered that a tumour is not a single mass of identical cells. Instead, it is made up of multiple, closely related groups of cancer cells that differ genetically and continue to evolve over time. As shown in Figure 2, these genetic differences appear at multiple levels:
- Within one tumour (intra-tumoural heterogeneity);
- Between separate metastases in the same patient (inter-metastatic heterogeneity);
- Within a metastasis (intra-metastatic heterogeneity);
- Between patients with the same cancer type (inter-patient heterogeneity).
As a result, different tumours can respond very differently to the same therapy. This helps explain why modern cancer care increasingly relies on drug combinations rather than single regimens. However, these strategies still fall short of dose personalisation. Drug combinations remain rooted in the single “best dose” paradigm and thus cannot account for patient-to-patient differences or for how drugs interact when combined. Addressing this problem will require a different way of thinking about how cancer therapies are tested and optimised.
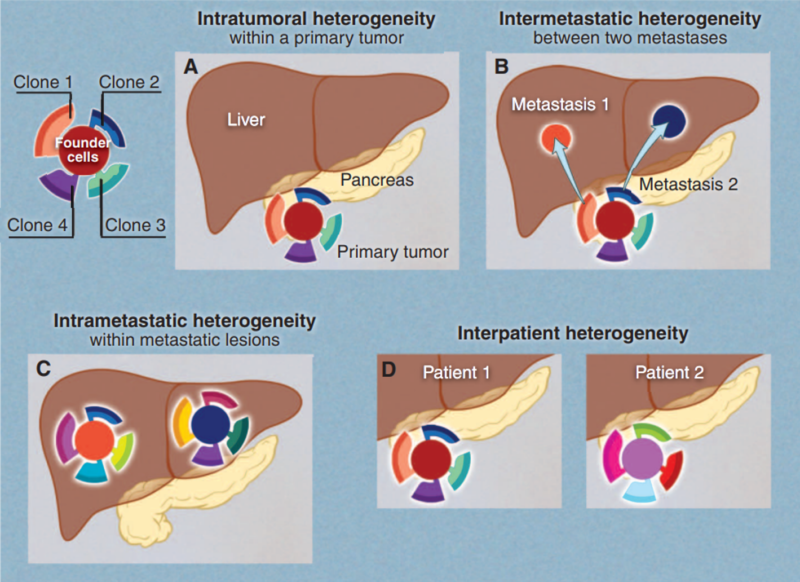
Figure 2. Tumour heterogeneity across space and patients. Pancreatic cancer with liver metastasis is used as an example. Cancer typically contains genetically different subpopulations or clones, contributing to various forms of heterogeneity. (A) Intra-tumoural: different regions within a tumour can contain distinct clones. (B) Inter-metastatic: separate metastatic tumours in the same patient may originate from different subclones and differ genetically. (C) Intra-metastatic: even within a single metastasis, multiple subclones can coexist and continue to evolve. (D) Inter-patient: tumours from different patients with the same cancer type can have different genetic profiles. Source: Vogelstein et al. (2013), Science.
Rationale of the I-PREDICT Trial
To address these limitations, Jason Sicklick, MD, FACS, a professor of surgery, and colleagues at the University of California, San Diego, U.S., conducted the Investigation of Profile-Related Evidence Determining Individualised Cancer Therapy (I-PREDICT) trial. Unlike traditional trials that assign patients to predefined treatment arms, I-PREDICT follows a different premise: each patient would receive a therapy regimen tailored to the specific molecular profile of their own tumour, with both drug selection and dosing customised accordingly.
Newly published in the Journal of Clinical Oncology, I-PREDICT enrolled patients with lethal advanced or metastatic cancers who had exhausted standard therapy options. These patients thus represented some of the most clinically challenging cases. All patients underwent genetic sequencing of tumour tissue and/or circulating tumour DNA. A multidisciplinary tumour board reviewed these molecular profiles and recommended personalised combinations of drugs approved by the Food and Drug Administration (FDA). These include immune checkpoint inhibitors (a type of immunotherapy), targeted agents (e.g., antibody-drug conjugates or small-molecule inhibitors), chemotherapy drugs and hormonal agents (Figure 3).
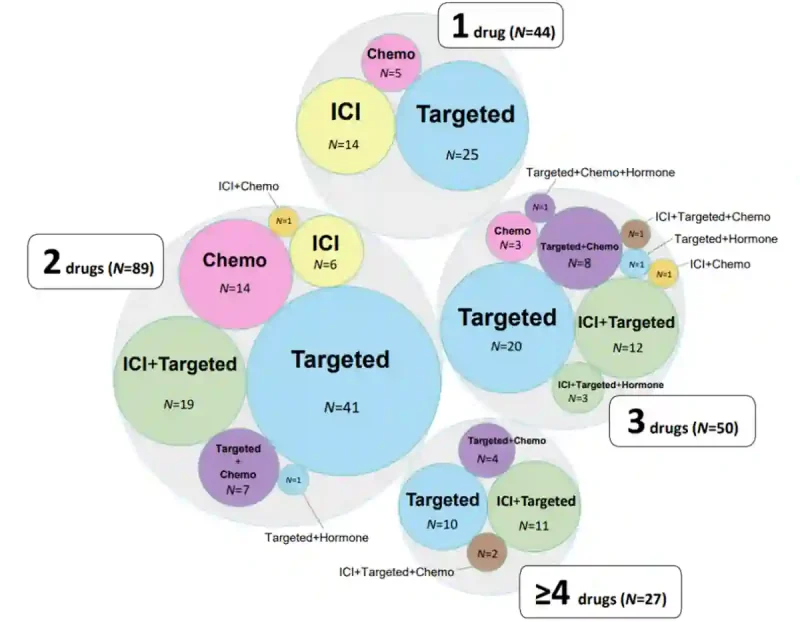
Figure 3. Diversity of therapy combinations in the I-PREDICT trial, combining targeted agents, immune checkpoint inhibitor (ICI), chemotherapy and hormonal therapy in various configurations. Most patients received two-drug combinations (N = 89), followed by three-drug (N = 50), single-drug (N = 44) and four-drug and above (N = 27). Source: Sicklick et al. (2026), Journal of Clinical Oncology (in supplementary material).
Interestingly, most combinations were used off-label, meaning the drugs were prescribed outside their original indications (i.e., what they were approved for). Many of the combinations had also never been tested in phase I trials, where safety, dosing limits and drug interactions are normally established. In fact, given the enormous number of possible drug-drug-dose permutations, it would be impractical to conduct phase I trials for each potential combination. With approximately 300 cancer drugs available for clinical use, more than 45,000 possible two-drug combinations and over 4.5 million three-drug combinations can be generated.
To navigate this challenge, Prof. Dr Sicklick et al. adopted a different dosing strategy. Instead of the traditional inter-patient (between patients) dose-escalation model used in phase I trials, I-PREDICT implemented intra-patient (within a patient) dose optimisation. Patients receiving novel drug combinations were started at reduced initial doses, followed by careful dose escalation or reduction based on individual tolerance and clinical response. In effect, dose finding occurred within each patient (intra-) rather than across a group of patients (inter-).
This approach allowed the study to move beyond the rigid framework of conventional drug development, generating new therapeutic insights while maintaining safety oversight. As Dr. Sicklick and colleagues noted, “We hypothesized that optimised personalised precision cancer medicine is not just about matching patients to the best medication(s) for their tumours, but also about safely administering the right drugs and doses to patients, including previously unstudied combinations.”
Results of the I-PREDICT Trial
After recruitment and treatment, 210 evaluable cancer patients remained. About 95% of them harboured distinct tumour genomic profiles, highlighting the extent of tumour heterogeneity encountered in clinical practice. In this cohort, 157 distinct therapy regimens were administered, including 103 combinations that had not undergone phase I testing.
Despite the use of these novel combinations, rates of severe therapy-related toxicity were low and were, in fact, lower than among patients receiving established regimens with fixed doses. This likely reflects the conservative intra-patient dose adjustment strategy, in which dose reductions were more frequent than escalations. This allows therapy intensity to be personalised according to tolerability rather than dictated by fixed starting doses.
With safety established, Sicklick et al. then evaluated clinical outcomes. Encouragingly, the degree to which therapies matched a patient’s tumour profile – quantified by a molecular matching score (MS) – showed a strong association with progression-free survival (PFS), overall survival (OS) and disease control rates (DCR). Specifically, patients were stratified into five groups according to their MS: i.e., 0%, 1–39%, 40–59%, 60–99% and 100%. Clinical outcomes improved progressively across these groups, with MS correlating strongly with PFS (R² = 0.76), OS (R² = 0.94) and DCR (R² = 0.81) (Figure 4).
(Note: The R² value reflects the strength of the relationship between MS and clinical outcomes, with values closer to 1 indicating a stronger association.)
To better appreciate this, we can look at the clinical differences between the extremes. Patients with 0% matching achieved a median PFS and OS of 2.4 and 9.1 months, whereas those with 100% matching achieved a median PFS and OS of 10.4 and 21.2 months, respectively. DCR similarly increased from 23.3% in the unmatched group to 71.4% in the fully matched group (Figure 4). Put simply, the more completely a therapy regimen matches the specific genetic alterations of the patient’s tumour (higher MS), the better the patient fared.
These outcomes were not influenced by the number of drugs administered or dose intensity. This means that simply giving more drugs at higher doses did not translate into clinical benefit. It was not about more treatment but better-matched treatment (quality over quantity). The precision of drug matching and dose tolerability were the main drivers of benefit.
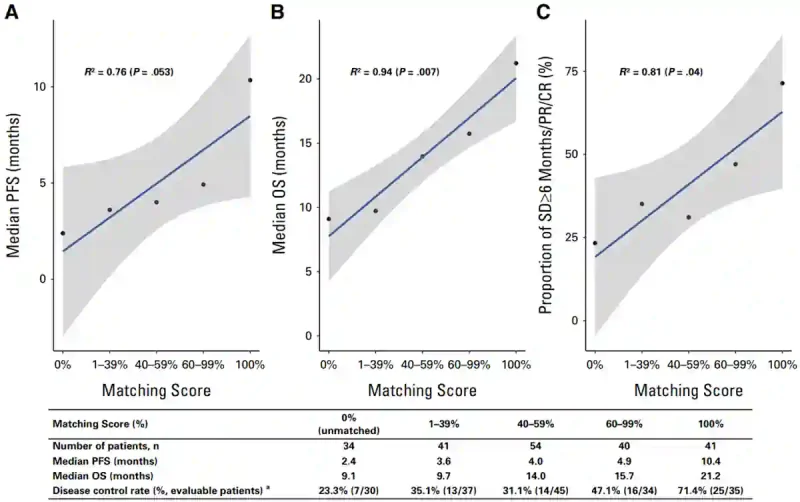
Figure 4. Molecular matching score (MS) correlates with clinical outcomes in I-PREDICT. Patients were stratified into five cohorts based on the proportion of tumour alterations targeted by therapy. Higher MS were associated with progressively improved progression-free survival (PFS), overall survival (OS), and disease control rates (DCR). Patients with 100% molecular matching experienced substantially longer survival than those receiving unmatched therapy. Source: Sicklick et al. (2026), Journal of Clinical Oncology.
Conclusion of the I-PREDICT Trial
Cancer is biologically heterogeneous, yet drug development has long relied on standardised therapy models built for averages rather than individuals. The pioneering I-PREDICT trial suggests that the future of precision oncology may depend less on discovering new drugs and more on rethinking how existing ones are combined and dosed. Even the FDA is starting to align with this philosophy through initiatives such as Project Optimus, which seeks to reform dose optimisation in oncology by recognising that the traditional “more is better” paradigm often produces excess toxicity without added efficacy. As Dr. Sicklick said in a press release, “Instead of a one-size-fits-all, we are moving toward one-size-fits-one.”

